|
COORDINATOR: LUISA LANFRANCO
The main goals of the workpackage are i) to provide the annotation of the Candidatus Glomeribacter gigasporarum genome, that is, to give a functional classification of all the identified genes; ii) to explore the potentials of this completely unknown microbe as a “genes reservoir”, that could be considered as a source of metabolic pathway and possibly novel bio-active molecules of biotechnological interest; iii) to develop a data warehouse dedicated to Ca. G. gigasporarum oriented to the management of genomic sequences, annotation, functional and comparative data.
Genome annotation is a rapidly evolving field in genomics made possible by the largescale generation of genomic sequences and driven predominantly by computational tools. The goal of the annotation process is to assign as much information as possible to the raw sequence of complete genomes with an emphasis on the location and structure of the genes. This can be accomplished by ab initio gene finding, by identifying homologies to known genes from other organisms, by the alignment of full-length or partial mRNA sequences to the genomic DNA, or through combinations of such methods.
Genome annotation currently depends on discovering homologies to genes with known functions from other species and thus strongly relies on comparative genomics studies. At large scale comparative genomics is the analysis and comparison of genomes from different species.
The genome annotation is a crucial step towards the identification and the characterization of genes involved in specific metabolic pathways. Bioinformatics analyses give predictions of gene function; this information is instrumental to plan targeted experiments to finally confirm the biochemical activity and/or the biological role of given DNA sequences.
Alignment of DNA sequences is the core process in comparative genomics. Several powerful algorithms have been developed to align two or more sequences. Some of these sequence-similarity tools are publicly accessible over the Internet (e.g. BLAST). However, the computational power required to align billions of nucleotides between two or more species vastly exceeds what is normally available. Thus, several research groups make available pre-computed alignments of genomes through servers or browsers. To this respect, the relevance of building tools that allow to “navigate” flexibly data, from arbitrary, at least in principle, perspectives, under different degrees of approximation, is rapidly growing. These requirements match the typical design and implementation principles of data warehouse, a concept that dates back at least to the mid-1980s. Basically, since its introduction it was meant to provide architectural and conceptual models for exploiting data and information, extracted from a set of traditional databases, inside tools able to support people-involved decisional tasks. The systematic support to the organization and storage in large structures of large-scale collections of data, the related ability to carry out extensive, multifaceted querying by means of a data warehouse that uses protocols harnessing data stored remotely in distinct databases, and the possibility to be queried them flexibly, are gaining evidence as key factors for the successful development of tools to explore genetic and genomic relatedness. Some examples of databases developed in the area of bioinformatics are ENZYME, KEGG, BioCyc, Universal Protein Resource (UniProt). A successful experience was recently described by Cornell et al. (2007), who presented the e-Fungi data warehouse. This tool was exploited to answer some fundamental questions about fungal evolutionary biology, including patterns of gene loss and gene duplication and extent of protein family conservation among the kingdom Fungi, and enable integrative analyses of genome sequence and functional genomic data.
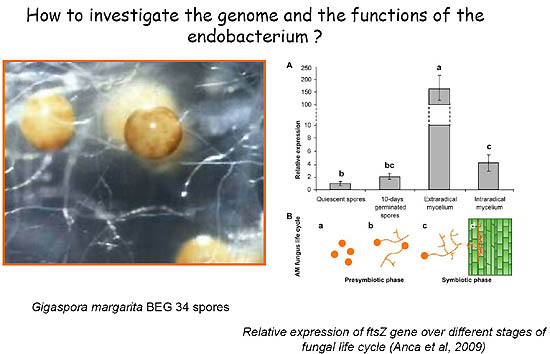
|
